Die Zulassung von Tierarzneimitteln führt durch umfangreiche Prüfungen – Produkte unterliegen einer kontinuierlichen Nutzen-Risiko-Bewertung
 Qualität, Wirksamkeit und Sicherheit zeichnen Tierarzneimittel aus. Die Hürden für eine Zulassung sind entsprechend hoch. Der Blickpunkt sprach mit Dr. Sabine Schneider, Novartis Tiergesundheit GmbH und Vorsitzende der Arbeitsgruppe Zulassungsfragen des Bundesverbandes für Tiergesundheit (BfT), über den Zulassungsprozess bei Tierarzneimitteln.
Qualität, Wirksamkeit und Sicherheit zeichnen Tierarzneimittel aus. Die Hürden für eine Zulassung sind entsprechend hoch. Der Blickpunkt sprach mit Dr. Sabine Schneider, Novartis Tiergesundheit GmbH und Vorsitzende der Arbeitsgruppe Zulassungsfragen des Bundesverbandes für Tiergesundheit (BfT), über den Zulassungsprozess bei Tierarzneimitteln.
Blickpunkt: Wie lange dauert es, bis aus einem neuen Wirkstoff ein zugelassenes Tierarzneimittel wird?
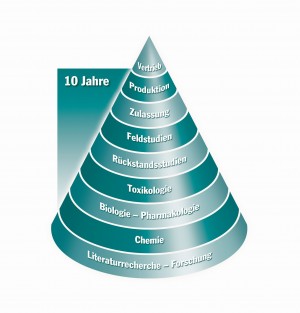
Dr. Schneider: Ein Tierarzneimittel erhält von den Behörden die Zulassung für den deutschen bzw. europäischen Markt, wenn der pharmazeutische Unternehmer belegen kann, dass das Produkt sicher ist für Mensch, Tier und Umwelt. Grundlagen für die Zulassung eines Tierarzneimittels und eines Humanarzneimittels sind das deutsche Arzneimittelgesetz und zahlreiche europäische Verordnungen und Richtlinien. Wir gehen heute davon aus, dass zwischen der Entdeckung eines neuen Wirkstoffes und dem Verkauf des fertigen Tierarzneimittels bis zu zehn Jahre liegen. Die Kosten für die Neuentwicklung eines Tierarzneimittels bis hin zur Marktreife liegen nach Schätzungen bei bis zu 150 Millionen Euro.
Blickpunkt: Welche Schritte müssen dabei im Detail durchlaufen werden?
Dr. Schneider: Die Zulassung selbst, d. h. die Einreichung der Dokumentation bei den Behörden bis zum Erhalt des Zulassungsbescheides dauert heute circa ein bis zwei Jahre. Vorab sind allerdings die Experten in den Forschungsabteilungen der Unternehmen damit beschäftigt, Studien durchzuführen und Daten zusammenzutragen, die dann im sogenannten Zulassungsdossier bei den Behörden eingereicht werden. In Deutschland bewertet das Bundesamt für Verbraucherschutz und Lebensmittelsicherheit in Berlin die eingereichten Unterlagen. Für Tierimpfstoffe, auf die ich hier nicht näher eingehe, ist das Paul-Ehrlich-Institut zuständig.
Das Zulassungsdossier besteht aus mehreren Teilen. Im Teil I befinden sich der Zulassungsantrag, Expertengutachten sowie die Vorschläge für die Kennzeichnung, Packungsbeilage und für die Zusammenfassung der Produkteigenschaften. Teil II bezieht sich auf die chemisch pharmazeutische Qualität, hier belegt der pharmazeutische Unternehmer die Qualität aller Inhaltsstoffe und des Fertigproduktes. Er beschreibt die Herstellung sowie die Kontrollen der einzelnen Herstellungsschritte. Auch die Haltbarkeit des Produktes ist zu belegen.
In Teil III geht es nochmals besonders um die Sicherheit für Mensch, Tier und Umwelt. Hier werden Studien zur Pharmakologie und Toxikologie dargelegt. Ein besonderes Augenmerk wird bei Tierarzneimitteln auf die Rückstandsstudien und die Umweltdaten gelegt. Teil IV befasst sich mit der Wirksamkeit des Produktes. Aufgrund dieser Daten erfolgt auch eine Nutzen-Risiko-Abwägung. Ein Tierarzneimittel sollte eine möglichst gute Wirksamkeit aufweisen, aber möglichst wenige Nebenwirkungen zeigen.
Blickpunkt: Welche Vorteile bringt eine europäische Zulassung gegenüber der nationalen?
Dr. Schneider: Seit mehr als 15 Jahren gibt es europäische Zulassungsverfahren. Ziel war es damals, die Zulassungsprozesse europaweit zu harmonisieren und zu vereinfachen. Dies wurde in einigen Bereichen auch erreicht. Mit Hilfe eines zentralen Zulassungsverfahrens für innovative Produkte erhält der pharmazeutische Unternehmer eine europäische Zulassungsnummer und kann nach erfolgreicher Zulassung das Tierarzneimittel in allen europäischen Ländern vertreiben.
Bei anderen europäischen Verfahren werden zwar weiterhin nationale Zulassungen ausgesprochen, diese beruhen allerdings auf einem gemeinsamen europäisch koordinierten Bewertungsverfahren. Weiterhin arbeiten aber alle europäischen Zulassungsbehörden an der Bewertung des Dossiers.
Blickpunkt: Verschwindet ein einmal zugelassenes Tierarzneimittel aus dem Fokus der Kontrollmechanismen?
Dr. Schneider: Auch nach der Zulassung sind wir als pharmazeutischer Unternehmer verpflichtet, die Zulassung immer auf dem aktuellen Stand zu halten. Dies bedeutet, dass beispielsweise Änderungen bei der Herstellung oder neue Erkenntnisse zur Sicherheit oder Wirkung des Produktes den Behörden angezeigt werden. Gebrauchsinformationen und Verpackungstexte werden regelmäßig angepasst.
Die Kosten für die Zulassung haben sich in den letzten Jahren aufgrund der gestiegenen Anforderungen deutlich erhöht. Nicht zu unterschätzen sind die Erhaltungskosten, die mit 35 Prozent der Forschungs- und Entwicklungskosten in den EU Ländern deutlich höher liegen als in anderen Ländern weltweit.
Blickpunkt: Bestehen bezüglich des Zulassungsprozesses bei der Industrie noch offene Wünsche?
Dr. Schneider: Die Kompetenz der Zulassungsbehörden in den einzelnen EU Ländern ist hoch, eine sinnvolle Arbeitsteilung bei der Bewertung der Zulassungsunterlagen könnte jedoch noch viel Zeit und Geld einsparen.
Bereits heute wird an dem 1-1-1-Konzept gearbeitet. Dies bedeutet: ein Binnenmarkt, ein Zulassungsdossier, eine Zulassung in der EU. Auch für den Verbraucher würde hier die bestmögliche Qualität und Sicherheit im Vordergrund stehen.